FARFAR (RNA De Novo) Server Documentation
Overview:
The main RNA structure modeling algorithm in Rosetta is based on the assembly of short (1 to 3 nucleotide) fragments from existing RNA crystal structures whose sequences match subsequences of the target RNA. The Fragment Assembly of RNA (FARNA) algorithm is a Monte Carlo process, guided by a low-resolution knowledge-based energy function. The models can then be further refined in an all-atom potential to yield more realistic structures with cleaner hydrogen bonds and fewer clashes; the resulting energies are also better at discriminating native-like conformations from non-native conformations. The two-step protocol has been named FARFAR (Fragment Assembly of RNA with Full Atom Refinement). [Check the citations below for more information.] Here are some tips to use the server and to interpret the results.
Tips:
- Do a trial run first, and view the molecule in PyMOL or your favorite viewer. This is particularly important if you have multi-stranded motif -- check that the strands are separated, and that any specified Watson-Crick pairs are reasonably paired.
- Input and output PDB models have residues marked rA, rC, rG, and rU, due to historical reasons. If you supply a "standard" PDB file as a native reference model, it will be converted to this format automatically. PDBs with non-standard nucleotides (e.g,. dihydroxyuridine) will not be handled properly. If you run into issues, try the conversion yourself with this python script.
- This method has been demonstrated to reach atomic accuracy for small motifs (12 residues or less) -- the current bottleneck for some of these motifs and for larger RNAs is the difficulty of complete conformational sampling (as in other applications in Rosetta, e.g., protein de novo modeling). On-going work attempts to resolve this issue, but requires greater computational expense and a workflow ('stepwise assembly') that is not yet straightforward to implement on a public server. It is available in the main Rosetta codebase, however (see Sripakdeevong et al., citation below.)
- This code is particulary useful for noncanonical loops and multi stranded motifs which are capped by canonical Watson-Crick helices (see below for examples). You will have multiple strands, you should specify a secondary structure that connects strands to each other by at least some Watson-Crick pairs [advanced users can use 'params' files, described below].
- The server is limited to RNAs of 32 nucleotides or shorter. For larger RNAs, other algorithms are being developed that use pre-computed ideal helices and 'motifs'. This code is not yet available on the server, but availabe in the main Rosetta codebase (see Kladwang et al. citation below).
- As with most other modes in Rosetta, the final ensemble of models is not guaranteed to be a Boltzmann ensemble.
- These models will be most useful for loops & motifs where you don't already know the answer -- de novo modeling cases. For puzzles that are obviously homologous to existing solved structures, it makes much more sense to use comparative modeling programs like this to create reasonable models. [This is analogous to the situation for protein modeling tools where different methods are used for de novo vs. comparative modeling.]
Inputs:
You need to input:
- The sequence, from 5' to 3'. Typically in lower-case letters, but upper-case is acceptable and will be converted. Use space (' '), *, or + between strands.
- The secondary structure in dot-parentheses notation. This is optional for single-stranded motifs, but required for multi-strand motifs. Note that even if a location is 'unpaired' in the input secondary structure (given by a dot, '.'), it is not forced to remain unpaired -- it is free to do what it wants.
- "Native" structure [Optional]. Providing this will result in heavy-atom rmsd scores being calculated with respect to this input model (rather than to the lowest energy structure found). Note again that the native pdb should have residues marked rA, rC, rG, and rU; pdbs in standard format will be converted.
Here are some examples:
- A simple hairpin. Sequence is:
gggcgcaagccu
Secondary structure [optional] could be((((....))))
Note that we could also leave out the secondary structure and let Rosetta fragment assembly sample a lot of secondary structures.
- A string of four purine/purine pairs (check out PDB file 283D) (a 4x4 'internal loop'), with the following toplogy:
5'-CGAAAG-3' |****| 3'-GAAAGC-5'
The sequence input would be:cgaaag+cgaaag
This is a two-strand motif, so we must specify a secondary structure, which would be:(....(+)....)
- A pseudoknot. (check out PDB file 1L2X), with the following schematic topology:
5'-GCGCGG---C ||||| A GCGCCUGCC G ||| AACAAACGG-3'The sequencegcgcggcaccguccgcggaacaaacgg
The above is enough, since this is a single-strand motif. But if you want to ensure the stems, you can include a secondary structure like this:.(((((..[[[.))))).......]]]
Here the Watson/Crick stems are 'non-nested', so the second stem is written with square brackets to avoid ambiguity. You can specify up to a single non-nested stem currently. See below (advanced) for more complex topologies.
Interpreting Results
- The server shows pictures of the best-scoring models from the 5 best-scoring clusters from the run in rank order by energy. The clustering radius, by default is 2.0 A. Click on the [Model-N] link to download the PDB file.
- The server returns cluster centers (without pictures) for the next 95 clusters as well, as well as the top 20 lowest-energy structures. These may be valuable if you are filtering models based on experimental data.
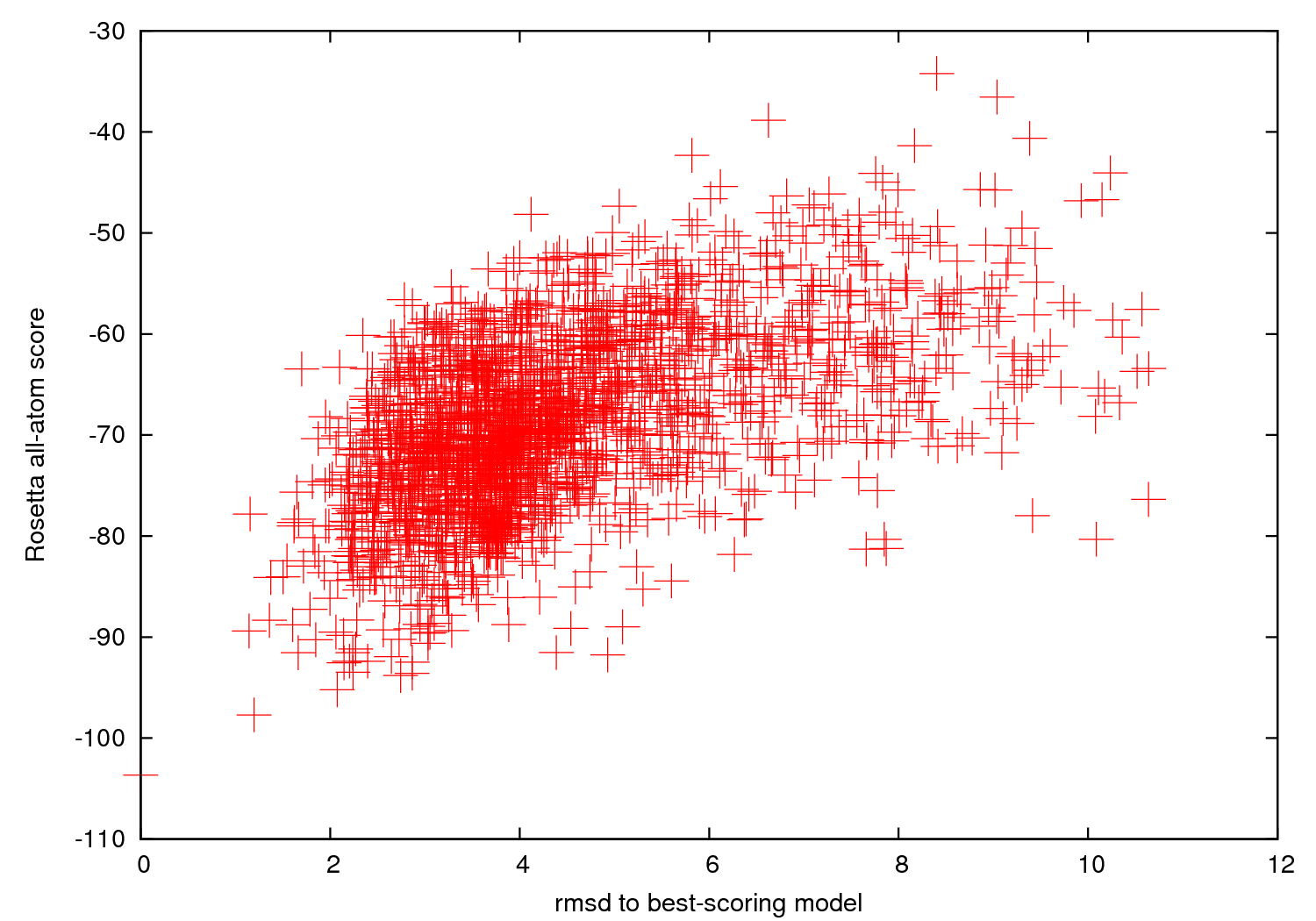
- The server also returns a 'scatter plot'
of the energies of all the models created. The x-axis
is a distance measure from your native/reference model -- here an rmsd (in Angstroms) over all heavy atoms. The y-axis is the score (energy) of the structure. In runs where you do not supply a native structure, the x-axis is a distance measure from the best scoring model found. As with nearly every Rosetta application, a hallmark of a
successful run is convergence, visible as an energetic "funnel" of low-energy structures
clustered around a single position.
- Finally you'll also get a score break down of all the models, which helps explain the energetics that go into Rosetta all-atom refinement for RNA:
That is, near the lowest energy model there are additional models within ~ 2 Angstrom RMSD, with an approximate correlation of score vs. RMSD. In such runs, the lowest energy cluster centers have a reasonable chance of covering native-like structures for the motif, based on our benchmarks (see Das et al., 2010, below).
A hallmark of an unsuccessful run is a lack of 'convergence' -- few structures within 2 A RMSD of the lowest energy model:

If you get this, you might consider running a bigger job (more models).
score Final total score
fa_atr Lennard-jones attractive between atoms in different residues
fa_rep Lennard-jones repulsive between atoms in different residues
fa_intra_rep Lennard-jones repulsive between atoms in the same residue
lk_nonpolar Lazaridis-karplus solvation energy, over nonpolar atoms
hack_elec_rna_phos_phos Simple electrostatic repulsion term between phosphates
ch_bond Carbon hydrogen bonds
rna_torsion RNA torsional potential.
rna_sugar_close Term that ensures that ribose rings stay closed during refinement.
hbond_sr_bb_sc Backbone-sidechain hbonds close in primary sequence
hbond_lr_bb_sc Backbone-sidechain hbonds distant in primary sequence
hbond_sc Sidechain-sidechain hydrogen bond energy
geom_sol Geometric Solvation energy for polar atoms
atom_pair_constraint Harmonic constraints between atoms involved in Watson-Crick base pairs
specified by the user in the params file
linear_chainbreak for 'temporary' chainbreaks, penalty term that keeps chainbreaks closed.
N_WC number of watson-crick base pairs
N_NWC number of non-watson-crick base pairs
N_BS number of base stacks
[Following are provided if the user gives a native structure for reference]
rms all-heavy-atom RMSD to the native structure
rms_stem all-heavy-atom RMSD to helical segments in the native structure, defined by 'STEM' entries in the parameters file.
f_natWC fraction of native Watson-Crick base pairs recovered
f_natNWC fraction of native non-Watson-Crick base pairs recovered
f_natBP fraction of base pairs recovered
Incorporating 1H NMR chemical shift data:
Please refer to Sripakdeevong et al. (PDF) for a detailed description of the ROSETTA chemical shift guided RNA denovo modeling method.
Instructions:
- Preparing chemical shift data file:
- The inputted chemical shift data file should be in STAR v2.1 format.
- Please include only chemical shift data lines.
- Our method use only non-exchangable 1H chemical shift data. Data lines belonging to other atom types will be ignored.
- Here is an example chemical shift data file for the tandem GA:AG mismatch motif: tandem_GA_1MIS.str.
- We recommend including two W.C. basepairs at every strand ends:
- We recommend selecting "Allow bulge" and "Use updated (2012) force-field" options to reproduce the results in Sripakdeevong et al. (PDF)
Example (tandem GA:AG mismatch):
Input sequence: cggacg+cggacg
Input secstruct: ((..((+))..))
Topplogy: 5'-CGGACG-3'
||**||
3'-GCAGGC-5'
Native PDB: tandem_GA_1MIS.pdb
Advanced:
Advanced users can also sequences and strand-division/secondary-structure information as fasta and params files, respectively. This is the file format that is used in current Rosetta command-line input, and allows for more flexibility in specifying non-Watson-Crick pairings, where to make cutpoints, etc.
- The fasta file has the RNA name on the first line (after >), and the sequence on the second line. Valid letters are a,c,g, and u:
> my favorite RNA sequence aaacccggguuu
Important note: that unlike above, this input does not accept +,* or space between nucleotides. You need to specify those strand breaks in the params file, described next.
- You can specify the bounding Watson/Crick base pairs, strand boundaries, and more in a "params file", with lines like
CUTPOINT_OPEN <N1> [ <N2> ... ] means that strands end after nucleotides N1, N2, etc. Required if you have more than one strand! STEM PAIR <N1> <M1> W W A [ PAIR <N2> <M2> W W A ... ] means that residues N1 and M1 should form a base pair with their Watson-Crick edges ('W') in an antiparallel ('A') orientation; and N2 and M2, etc.. One 'STEM' line per contiguous helix. This will produce constraints drawing the base-paired residues together, and will also provide potential connection points between strands for multi-strand motifs. OBLIGATE PAIR <N> <M> <E> <F> <O> means that a connection point between strands is forced between residues N and M using their edges E and F [permitted values: W ('Watson-Crick'), H ('Hoogsteen'), S ('sugar')] and orientation O [permitted values: A (antiparallel) or P (parallel), based on normal vectors on the two bases]. If modeling a single-strand motif, forcing this 'obligate pair' will result in a 'temporary' chainbreak, randomly placed in a non-stem residue. Typically will not use OBLIGATE except for complex topologies like pseudoknots. CUTPOINT_CLOSED <N> Location of a 'temporary' chainbreak in strands. Typically will not use except for complex topologies. CHAIN_CONNECTION SEGMENT1 <N1> <N2> SEGMENT2 <M1> <M2> Used instead of obligate pair -- if we know two strands are connected by a pair, but we don't know the residues, can ask for some pairing to occur between strands N1-N2 and M1-M2, sampled randomly. Typically will not use except for complex topologies.
Here are the examples described in the Inputs section above in fasta/params notation.
- The simple hairpin. Fasta file looks like this:
>1zih.pdb [GCAA tetraloop] gggcgcaagccu
We can also specify the closing stem with a params file like this (but this is not necessary for such a short motif):STEM PAIR 1 12 W W A PAIR 2 11 W W A PAIR 3 10 W W A PAIR 4 9 W W A
- The motif with four purine/purine pairs has this fasta file (concatenate the sequence of both strands, given in 5'-to-3' order):
>chunk001_283d_RNA.pdb cgaaagcgaaag
This is a two-strand motif, so we must specify a params file, showing that the first strand ends after nucleotide 6, and there should be Watson-Crick interactions between nucleotides 1 and 12, and 6 and 7:CUTPOINT_OPEN 6 STEM PAIR 1 12 W W A STEM PAIR 6 7 W W A
- The pseudoknot has fasta:
>1l2x_RNA.pdb gcgcggcaccguccgcggaacaaacgg
The above is 'enough', since this is a single-strand motif. But if you want to ensure the stems, you can include a params file like:OBLIGATE PAIR 11 25 W W A PAIR 10 26 W W A PAIR 9 27 W W A OBLIGATE PAIR 2 17 W W A PAIR 3 16 W W A PAIR 4 15 W W A PAIR 5 14 W W A PAIR 6 13 W W A
Again, you can try runs with and without the params file. - Advanced. Tetraloop/receptor tertiary contact connected by non-canonical base pairs. This involves a connection between a GAAA tetraloop at the end of one helical stem that docks into its 11-nt 'receptor' sequence ensconced within another helix.
5' 3' 5' 3' G--C C--G G A U U AA.....AA A G--U 5' 3'The motif involves three strands that connect into 3 helices.>chunk006_2r8s_RNA.pdb ggaaaccuaaguaug
Note that there are no Watson/Crick pairing from the tetraloop strand to either of the two strands in the receptor. If you happen to know that there is a non-canonical pairing from A3 to A13 involving their watson-crick edges, and putting those adenosines in a parallel arrangement, you could use the following params file:CUTPOINT_OPEN 6 11 STEM PAIR 1 6 W W A STEM PAIR 7 15 W W A STEM PAIR 11 12 W W A OBLIGATE PAIR 3 13 W W P
But if you didn't know that pairing, you can use the following:CUTPOINT_OPEN 6 11 STEM PAIR 1 6 W W A STEM PAIR 7 15 W W A STEM PAIR 11 12 W W A CHAIN_CONNECTION SEGMENT1 1 6 SEGMENT2 7 15
Note that neither params file can currently be encoded in the default secondary structure framework -- a params file is necessary.
More questions?
We welcome scientific and technical comments on our server. For support please contact us at Rosetta Forums with any comments, questions or concerns.