PeptiDerive identifies for a given protein-protein interaction the linear peptide segment that contributes most to binding. Such peptides may serve as candidates for modulation of the protein-protein interaction. As input, the user provides a structure of a protein complex. PeptiDerive provides as output a list of the peptides that contribute most significantly to the interaction, and presents quantitative and visual data about the binding mode and energetics.
Peptiderive Server Documentation
Algorithm

The protocol accepts a structural model of an interaction between a pair of protein molecules. The user specifies which of the proteins is considered to be the partner (the other termed here a receptor). A short minimization of the structure is performed using Rosetta to remove local clashes without changing the structure significantly. Then, a sliding window (of length defined by the user) goes over the partner chain. At each window position, the peptide fragment is isolated from the rest of the protein, and the binding energy is evaluated (after adding terminal charges). Fragments which constitute a significant portion of the binding energy are considered to be candidates for competing with the existing interaction. The peptide with the most significant binding energy is reported.
Anticipating that inhibitory peptides might need to be further stabilized, we calculate whether each peptide fragment may be cyclized by introduction of a disulfide bridge, i.e. where the amino acids flanking the original fragment (the leading and trailing amino acids around the sliding window) are near enough in space so that a mutation to cysteine can generate a disulfide bridge. When the binding energy of the peptide is significant (≥35% of the total binding energy of the protein-protein complex), we model the peptide with its flanking amino acids mutated to cysteines (resulting in a peptide that is two amino acids longer than the requested length), and minimize the bound peptide-receptor complex.
Inputs
Structures of protein-protein complexes may be submitted for processing by PeptiDerive using the submission form. The structure is either uploaded in a PDB-format file, or by specifying a PDB ID.
If not stated otherwise, the protocol will assume every chain may act both as a receptor and a partner in the context of an interaction, and search for dominant segments in each of the protein chains. However, the user may restrict the role of the chains so that the protocol considers them only as receptors, or only as partners. The user may specify one or more peptide window sizes. Finally, as in any task submitted to the ROSIE server, the user may attach a description, comments, and choose whether or not to make the inputs and outputs publicly available.
To avoid parsing problems, it is advisable to upload your own
PDB-format cleaned-up copy of the protein complex structure: make sure only
ATOM and TER records are present (specifically, avoid
including HETATM records) and remove any alternative conformations,
or zero-occupancy ATOM records. Inputs not conformant with these
guidelines might produce errors or unexpected results.
Example run 3GXU: Enter 3GXU (Crystal structure of
Eph receptor and ephrin complex) in the ‘PDB ID’ field, and
click the ‘Fetch PDB’ button. The PDB-format file is fetched from
the RCSB server and used as-is. Enter a short description of the job in the
‘Job short description’ field at the top of the form (e.g. Eph
receptor and ephrin complex). Finally, press the Submit button at the bottom of
the form to queue the job for processing, leaving all other settings
untouched.
The results for 3GXU are available here for your inspection and described in
detail in the following.
Results
The results page provides a visual representation of the job inputs and outputs, as well as links where models and data files may be downloaded. All visualizations of proteins are generated using the PyMOL program (http://www.pymol.org/).
The topmost ‘Inputs’ section includes a figure of the submitted structure, and a link to the input PDB file. To the right, the ‘Status’ section shows details about the job, such as whether the job is queued, running or complete, and includes the parameters specified when the job was submitted, such as the description, restriction to receptor/partner chains and requested peptide window lengths.
Once the job is completed, an ‘Output’ section will appear below the ‘Inputs’ section. There you will find three subsections, ‘Derived peptides’, ‘Raw report file’ and ‘Scores’.
The ‘Derived peptides’ subsection provides visualization of the peptides derived from each receptor-partner complex, and their binding energies. The results for each receptor-peptide complex and window size (peptide length) are displayed in a separate tab. The peptide contributing most to the interaction energy is highlighted in the context of the receptor-partner complex in an overview figure to the right, and to the left, a close-up figure shows the peptide in sticks within the context of the receptor surface. Links are provided to download locations of both models in PDB file format. Following these figures, a graph shows the energy contribution of the peptides at each sliding window start position. Positions matching peptides candidate for cyclization are highlighted. The graph is sectioned vertically into areas that correspond to different relative energy contribution ranges, to effectively evaluate the significance of different linear peptide stretches.
The ‘Raw report file’ subsection shows the output report produced by the PeptiDerive program (see algorithm), and a download link is provided. The interface score corresponds to the binding energy of the protein-peptide complex at the corresponding position, while the total interface score corresponds to the entire receptor-partner protein-protein complex. If present, the cyclized interface score corresponds to the binding energy of the cyclized peptide and the receptor.
The ‘Scores’ subsection at the bottom shows the scores produced by Rosetta, broken down by score term, for each of the models produced, including protein-protein and protein-peptide complexes. For protein-peptide complexes, we use the following nomenclature to distinguish between four possible protein-peptide models: ‘best_linear’ or ‘best_cyclic’ corresponds to the peptide window position that scored best among the linear or cyclization-candidate peptides; the adjacent ‘linear_peptide’ or ‘cyclic_peptide’ suffix designates if this is the peptide as it was cut out from the partner protein (linear), or whether it is the cyclized model, if such a model was produced.
Finally, at the bottom of the page are download links, leading to all the generated input and output files of the job. Among these are log files of the PeptiDerive program, which may be useful for troubleshooting.
Interpreting results
Following the calculation of PeptiDerive, one may question what is the ability of the candidate peptide to bind its target and to interfere with the original interaction. One heuristic measure to assess the binding ability is to compare the interface energy as evaluated by Rosetta (see Binding energy) of the candidate peptide to the distribution of interaction energies as calculated for a dataset of solved structures of peptide-protein interactions (peptiDB; diamonds in Figure 1).
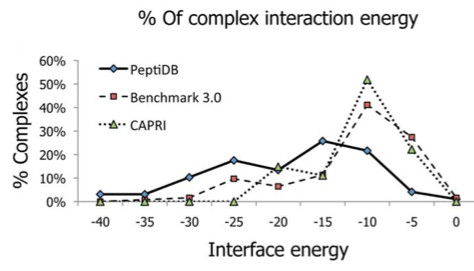
Figure 1: Calculating the absolute interaction energy values of the highest-affinity peptides of length 10 (from two different datasets - Benchmark 3.0 and CAPRI) Energies given in Rosetta energy units; REU), showed that a large number of the proposed peptides have energetic values that are comparable to known peptide interactions.It is shown that a significant fraction of the peptides derived from linear segments with the highest affinity contribute more than -10, and in some cases even more than -15 REU. Such values overlap with interaction energies typically calculated for native peptide-protein interactions in the peptiDB dataset.
A second desired feature in an inhibitory peptide, is the retention of the original binding mode, to allow competitive inhibition of the protein it was derived from. Namely, inhibitory peptides are expected to lie in the minimum of an energy funnel in their original binding pockets. Indeed previous benchmarking has shown that computationally derived peptides (the result of a Peptiderive calculation) favor the native binding conformation observed in the full protein.
FlexPepDock is a high-resolution peptide modeling protocol that is designed to dock peptides to their receptor with full peptide backbone flexibility and side-chain modeling, starting from an initial description of the complex. It is possible to use the FlexPepDock modeling protocol to aggressively sample the energy landscape of the derived peptide around the original binding pocket. Doing so allowed us to show that derived peptides favor the native binding conformation observed in the full protein.
For each derived peptide, we created 9,000 models of its complex with the cognate target of its origin domain. The different models cover a wide range of peptide conformations around the original binding pocket, deviating up to well over 15 Angstrom backbone RMSD from the original native structure.
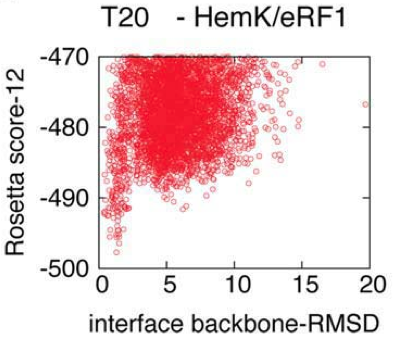
Figure 2: The plot above illustrates the energy landscape of a representative peptide derived from the CAPRI dataset, describing a distinct funnel with a near-native energy well. Each model is presented as a dot, indicating its total energy (y-axis) vs. the backbone RMSD of the peptide interface residues to the peptide in its native protein context (x-axis). For clearness, only top-scoring models out of the total 9000 are shown.
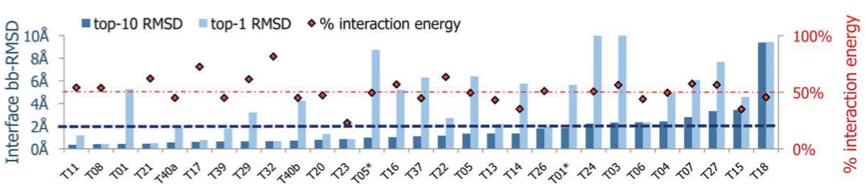
Figure 3: Overall, in most of the chosen representative interactions (72%), a near-native conformation (below 2 Angstrem backbone-RMSD over peptide interface residues) was found among the 10 top-scoring models, and in 35% of the interactions, a near-native conformation was even obtained by the top-scoring model, as shown in the plot above. Mapping the energy landscapes of peptides derived from CAPRI targets indicates that derived peptides favor the original binding conformation for a large number of targets. For each interaction, the interface backbone RMSD of the lowest-energy model created for the derived peptide is shown (dark blue bar), as well as for the best out of the 10 top scoring models (light blue bar). Red diamonds indicate for each target the fraction of binding energy that the derived peptide contributes (a dotted line is drawn at 50%).
Using the FlexPepDock app (to be incorporated in ROSIE as well), one can easily assess the local binding energy landscape of an inhibitory peptide suggested by Peptiderive.
Questions and answers
-
What input is accepted?The app expects PDB input. To avoid parsing problems, make sure your PDB file is made up of only
ATOMandTERrecords, i.e. no heteroatoms (HETATM) are included, and that the occupancy column is filled (no double conformations or 0.00 occupancy) -
Can I include modified residues?
To use phosphorylated residues, make sure that:
- they are named correctly (i.e. TYR/SER/THR and not PTR/SEP)
- modified residue coordinates (as well as the phosphates) are included as ATOM records (rather than HETATM)
Below you can find an example as to how a phospho-serine should look like:
ATOM 5776 N SER B 10 -19.024 43.939 120.740 1.00 0.00 ATOM 5777 CA SER B 10 -20.442 43.615 120.653 1.00 0.00 ATOM 5778 C SER B 10 -20.869 42.699 121.792 1.00 0.00 ATOM 5779 O SER B 10 -20.125 41.804 122.194 1.00 0.00 ATOM 5780 CB SER B 10 -20.750 42.972 119.314 1.00 0.00 ATOM 5781 OG SER B 10 -22.089 42.569 119.219 1.00 0.00 ATOM 5782 P SER B 10 -22.461 41.858 117.817 1.00 0.00 ATOM 5783 O1P SER B 10 -24.008 41.465 117.873 1.00 0.00 ATOM 5784 O2P SER B 10 -21.536 40.564 117.675 1.00 0.00 ATOM 5785 O3P SER B 10 -22.170 42.912 116.653 1.00 0.00
-
Can I include small molecules?At the moment the app can not handle small molecules at the interface. We intend to implement this in future releases.
-
How long do jobs take to complete?Running time depends on the size of the protein, the number of chain pairs in the proteins, and on the queue. Peptiderive takes only a few minutes to complete calculations on most proteins in the PDB.
-
What are the figures focusing on?Figures are created automatically by a PyMOL script, with the intention of focusing on the complex from the most informative angle. Since they are automatic, they sometimes fail. We encourage you to download the models and view them in your favorite molecular viewer.
-
Why is there no output for certain chain pairs?No output will be produced if the peptide you asked to derive is longer than the chain from which you intend to derive it. For example, if you requested to derive a peptide of length 9 from chain B, while the length of chain B length is 7 residues in total, the protocol will return no result. Note that the default length of the peptide to derive is 10 amino acids.
We welcome scientific and technical comments on our server. For support please contact us at Rosetta Forums with any comments, questions or concerns.
The ROSIE Peptiderive app was developed by Yuval Sedan at the Furman Lab, HUJI.