ROSIE Docking Server Documentation
Tips:
The RosettaDock Server performs a local
docking search. That is, the algorithm will search a set of
conformations near the given starting conformation for the
optimal fit between the two partners. Some suggestions:
-
You must upload a reasonable guess for the starting position. Place the protein partners near contact (but not overlapping) with the relevant patches of the proteins facing each other.
-
PyMol can be a useful tool to position proteins relative to each other. Use "editing" mode from the right panel, and try right-clicking to select a chain and "drag" to enable translation and rotation of the molecule (typically requiring left-shift + middle and left buttons). Finally from the main menu, File→Export Molecule can be used to write a PDB file containing the starting structure with both docking partners. The ‘align’ command may also be helpful if you are using a homologous complex as a guide.
Alternately, starting positions can be creating using one of several docking servers which perform global searches. Some leading servers include ClusPro, GRAMM-X, HEX, PatchDock, SymmDock, ZDOCK. Note that coordinate file formats from these servers might need to be brought into compliance for use by our server (e.g., putting a TER record between docking partners, assuring that the occupancy field is present and standard atom and residue names are used, etc.).
- Docking partners can be uploaded as two separate PDB files or a single PDB file. If there are multiple chains which serve as a single docking partner (e.g., the heavy and light chains of an antibody), the TER line should be removed between the two chains so that Rosetta knows to treat these as a single partner. Similarly, when creating a combined single input file with both partners, place a TER line between the two partners.
-
RosettaDock’s local perturbation includes ~ ±3 Å in the direction between the two proteins, ~ 8 Å in the directions sliding the proteins relative to each other along their surfaces, ~ 8° of tilt of the proteins, and a complete 360° spin around the axis between the centers of the two proteins. The server will start 1000 independent simulations from this range of random positions.
-
Given the local nature of the search, there is no need to include extra domains of the proteins beyond the two interacting domains. Trim unneeded residues out of your PDB file before uploading. The server will not accept PDBs larger than 600 residues total.
-
A fundamental assumption exploited by RosettaDock is that protein backbone conformations typically do not change much upon association. This holds for many proteins, but not all. If you believe that the backbone of one of your partners is flexible, you should be cautious with the results. For example, docking of a short, flexible peptide (~10 residues) is not likely to work, since a peptide lacks the tertiary interactions which stabilize full-size domains (75-250 residues). Similarly, docking will not capture the flexibility of a molecule like calmodulin; the correct backbone of the protein must be uploaded to begin. Docking of a single amino acid will not produce a reasonable result and is not allowed by the server.
- The PDB file format description can be found here.
- RosettaDock requires all backbone atoms to be present for any residue which appears in the starting structure (missing side-chains are acceptable since they will be rebuit). The error message "missing backbone
atoms" means there are one or more backbone atoms
missing in the input pdb file(s).
Interpreting Results
- The server returns the 10 best-scoring structures from the run in rank order by energy. Click on the [Model-N] link to download the PDB file (see below).
-
The server also returns a plot of the energies of all 1000 structures created. Each point on this plot represents a structure created by the server. The x-axis is a distance measure from your starting position, and the y-axis is the score (energy) of the structure. A hallmark of a successful run is an energetic "funnel" of low-energy structures clustered around a single position.
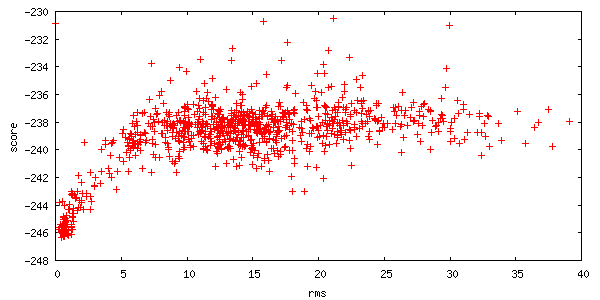
- For convenience, the full set of 1000 decoys is provided as compressed archive files. These decoys are not sorted or filtered.
-
For each predicted structure, a PDB file is given.
We welcome scientific and technical comments on our server. For support please contact us at Rosetta Forums with any comments, questions or concerns.
Happy docking!