RNA Redesign Server Documentation
Overview
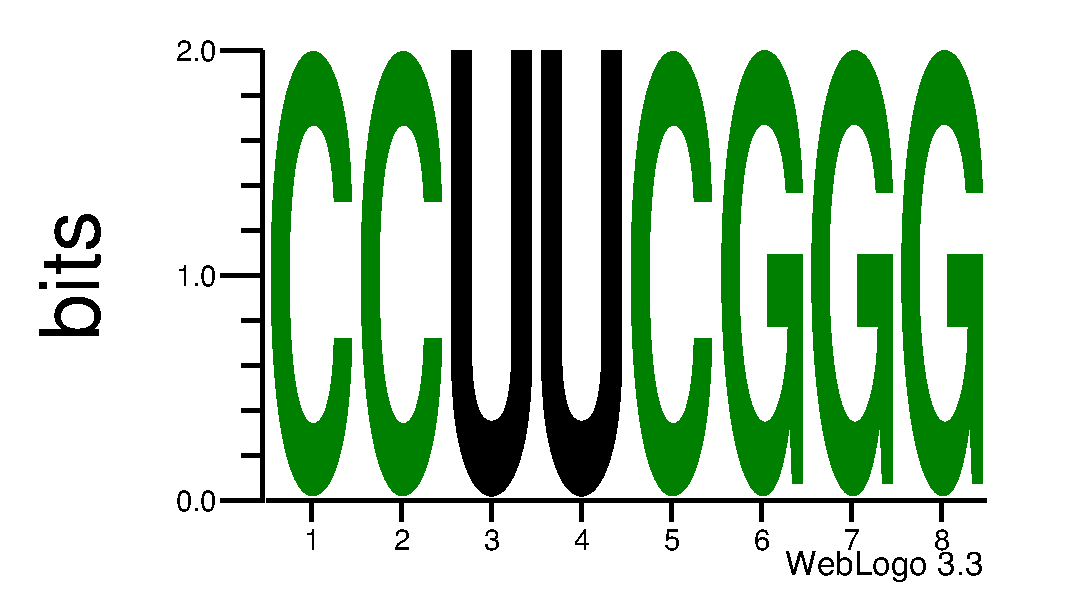
RNA Redesign performs fixed backbone design on RNA 3D structures to produce sequences that best stabilize a given 3D conformation. At each position being designed, each nucleotide is tried with set of different chi angles (the dihedral angle between the sugar and base). A summary of the sequences that stablize the submitted 3D structure are displayed in a weblogo to easy see the propensity of a sequence position to specific nucleotides. All designs are available to download and summerized in a table that can be sorted by Rosetta energy.Tips
- Input and output PDB models have residues marked rA, rC, rG, and rU, due to historical reasons. If you supply a "standard" PDB file as a native reference model, it will be converted to this format automatically. PDBs with non-standard nucleotides (e.g,. dihydroxyuridine) will not be handled properly. If you run into issues, try the conversion yourself with this python script.
- If there are only a few residues of interest, specify them in the optional text box. This will greatly speed up the run time.
Inputs
- PDB file: The pdb file of standard PDB format.
- Residues to design [Optional]: by default all residues will be designed, if there is only a section of the structure needs to be designed its possible to specify it. . Format: ChainName:FirstResidue-LastResidue, Example: A:90-100,B:2-4,C:10. In this case residues 90 to 100 on chain A, residues 2 to 4 on chain B and residue 10 on chain C will be designed while the rest will retain their native nucleotide.
- Number of requested designs: the number of designs to be generated, default (100), not necessarily unique from each other.
Interpreting Results
-
Displays a web logo to see the propensity of a nucleotide to a location in the 3D structure.
- The server shows pictures of the best-scoring sequences. Click on the [Model-N] link to download the PDB file
-
Finally you'll also get a score break down of all the models, which helps explain the energetics that go into Rosetta all-atom refinement for RNA
score Final total score fa_atr Lennard-jones attractive between atoms in different residues fa_rep Lennard-jones repulsive between atoms in different residues fa_intra_rep Lennard-jones repulsive between atoms in the same residue fa_elec_rna_phos_phos Simple electrostatic repulsion term between phosphates lk_nonpolar Lazaridis-karplus solvation energy, over nonpolar atoms ch_bond Carbon hydrogen bonds rna_torsion RNA torsional potential. rna_sugar_close Term that ensures that ribose rings stay closed during refinement. hbond_sr_bb_sc Backbone-sidechain hbonds close in primary sequence hbond_lr_bb_sc Backbone-sidechain hbonds distant in primary sequence hbond_sc Sidechain-sidechain hydrogen bond energy geom_sol Geometric Solvation energy for polar atoms
We welcome scientific and technical comments on our server. For support please contact us at Rosetta Forums with any comments, questions or concerns.
RNA modeling tools developed by the Das lab at Stanford