Symmetric Docking Server Documentation
The SymDock protocol predicts the structure of symmetric homooligomeric protein assemblies starting from the structure of a single subunit. It is very similar in spirit to the standard protein-protein docking protocol and contains the same basic components. The relative subunit orientation and conformations of side-chains are simultaneously optimized for a symmetric protein assembly. In contrast to the ROSIE docking server, the SymDock server does a global search for best docking orientation (5000 or 10 000 models, depending on symmetry type).
Inputs
The input is a monomeric subunit structure. The SymDock server currently supports two types of symmetries, cyclical (Cn) and dihedral (Dn) symmetry. The total number of subunits in the assembly has to be specified. With 4 subunits and cyclical symmetry that becomes a C4 symmetric tetramer, while 4 subunits with dihedral symmetry becomes a D2 tetramer. To limit computational cost no more than 10 subunits can be simulated with the server interface to SymDock.
Tips
- The server will not accept PDBs larger than 400 residues total.
-
A fundamental assumption exploited by RosettaDock is that protein backbone
conformations typically do not change much upon association. This holds
for many proteins, but not all. If you believe that the backbone of one of your
partners is flexible, you should be cautious with the results. Docking of a
single amino acid will not produce a reasonable result and is not allowed by
the server.
-
The PDB file format description can be found here.
Currently the PDB file can only
contain standard protein residues. You have to remove heteroatoms (such
as ligands) and convert selenomethionine residues to methionine etc. Some
PDBs may also have multiple conformations for each residue.
- RosettaDock requires all backbone atoms to be present for any residue which appears in the starting structure (missing side-chains are acceptable since they will be rebuit). The error message "missing backbone atoms" means there are one or more backbone atoms missing in the input pdb file(s).
Interpreting Results
-
The server returns the 10 best-scoring structures from the run in rank order by
energy. Click on the [Model-N] link to download the PDB file (see below).

-
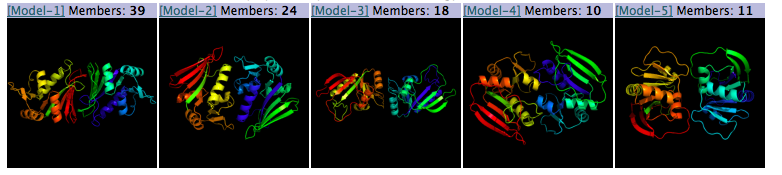
The server also returns the result of a cluster calculation on the top 400 models.
The algorithm divides the protein complexes into clusters, where each
member of the cluster is structurally similar (has an Ca rmsd of no larger
than 2 Å within the cluster). If we observe that the same interaction mode is
discovered several times during the simulation that is good sign. The model
at the center of the cluster for each of the largest clusters is returned. The
number of models in each cluster is printed above the image of the structure
for each cluster. Clustering results are only provided for systems with 6 or less
subunits.
-
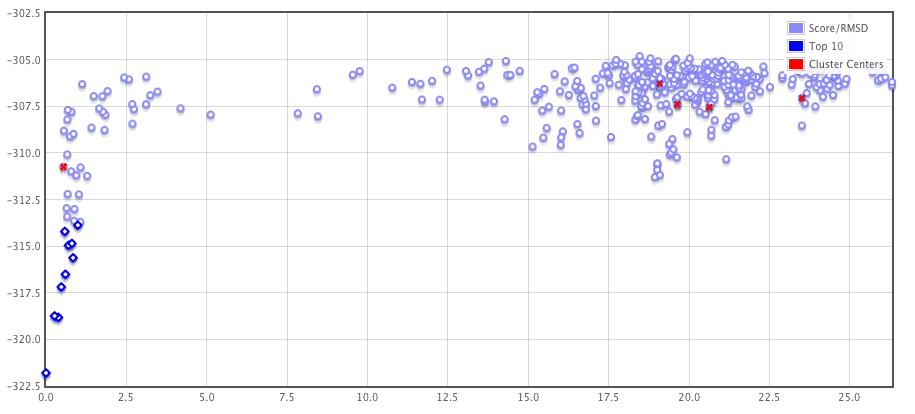
The server also returns a plot of the energies of the 400 lowest energy models.
Each point on this plot represents a structure created by the server. The y-
axis is the energy of the structure. The x-axis is a distance measure to a
reference complex structure (Ca rmsd in Angstrom). The reference complex
is the lowest energy model predicted by Rosetta. This is only true if the lowest
energy model is found among the models making up the top 5 clusters. If not
the reference model is selected to be the cluster center of the largest cluster.
The cluster centers for the 5 largest clusters are shown in the plot as well as
red points.
-
If there is an energy funnel towards the lowest energy structures (like in the case
shown here) that is a good sign. However, the lack of an energy funnel does
not necessarily mean that the correct binding mode is not found among the
400 predicted models. Often the correct binding mode can be found within a
cluster that does not have the lowest energy models but with many models. It
is also important that an the precence of an energy funnel is not a guarantee
that the correct complex structure was predicted.
- For convenience, the full set of the 400 lowest energy decoys, together with the top 5 cluster models, is provided as compressed archive files.
More information on how to run the SymDock application stand-alone can be found in:
DiMaio F, Leaver-Fay A, Bradley P, Baker D, André I. Modeling Symmetric Macromolecular Structure in Rosetta3 PLoS One. 2011;6(6):e20450. doi: 10.1371/journal.pone.0020450. link: http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0020450We welcome scientific and technical comments on our server. For support please contact us at Rosetta Forums with any comments, questions or concerns.
Modeling tools developed by André lab. The Rosie implementation was developed by Sergey Lyskov and Malte Johansson.